Case: GcnCat-01
|
You have this case description and two lab results due to von Gierke’s disease. What would you say about it?
6 year old boy with frequent episodes of weakness. Weakness accompanied by sweating, feeling of dizziness; weakness had been noticed since about age 4, but had become more noticeable when child entered school and was challenged by other children during recess. Enlarged abdomen, due to grossly enlarged liver. Kidneys also enlarged. Poor musculature. Normal heart. |

|
Fasting blood sample |
Pathology: Liver biopsy results |
|
Glucose
3.0 mmol/L (normal, 3.9 to 5.6) Lactate 7.1 mmol/L (normal, 0.56 to 2.0) Pyruvate 0.4 mmol/L (normal, 0.05 to 0.10) Free fatty acids 1.6 mmol/L (normal, 0.3 to 0.8) Triglycerides 3.0 g/L (normal, 1.5) Total ketone bodies 380 mg/L (normal, 30) pH 7.25 (normal, 7.35 to 7.44) Total CO2 12 mmol/L (normal, 24 to 30) |
Glycogen
10 g/ 100g of tissue (normal, up to 6 g) Lipid 20 g/100g of tissue (normal, less than 5 g) Glucose 6-phosphatase 20 units per gram of liver nitrogen (normal, 214 - 45) Phosphorylase 20 units per gram of liver nitrogen (normal, 22 - 3) Fructose 1,6-Bisphosphatase 9 units per gram of liver nitrogen (normal, 10 - 6) |
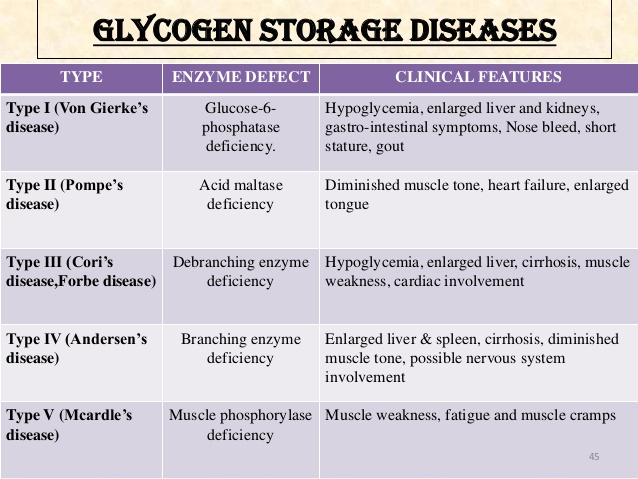
Synonyms of Glycogen Storage Disease Type I
- glucose-6-phosphatase deficiency
- glycogenosis type I
- GSDI
- Von Gierke disease
Subdivisions of Glycogen Storage Disease Type I
- glycogen storage disease type IA
- glycogen storage disease type IB
General Discussion
Glycogen storage diseases are a group of disorders in which stored glycogen cannot be metabolized into glucose to supply energy for the body. Type I glycogen storage disease is inherited as an autosomal recessive genetic disorder. Glycogen storage disease type I (GSDI) is characterized by accumulation of glycogen and fat in the liver and kidneys that can result in an enlarged liver and kidneys and growth retardation leading to short stature. GSDI is associated with abnormalities in the G6PC gene (GSDIA) or SLC37A4 gene (GSDIB) that result in enzyme deficiencies that cause excess amounts of glycogen accumulation in the body tissues and low levels of glucose in the blood. This enzyme deficiency also results in derangement of other important metabolites in the body thus causing imbalance or excessive accumulation of these metabolites.
Signs & Symptoms
The primary symptom of GSDI in infancy is a low blood sugar level (hypoglycemia). Symptoms of GSDI usually begin at three to four months of age and include enlargement of the liver (hepatomegaly), kidney (nephromegaly), elevated levels of lactate, uric acid and lipids (both total lipids and triglycerides), and seizures caused by repeated episodes of hypoglycemia. Continued low blood sugar can lead to delayed growth and development and muscle weakness.
High lipid levels can lead to the formation of fatty skin growths called xanthomas. Other conditions that can be associated with untreated GSD1 include osteoporosis, delayed puberty, gout (arthritis caused by accumulation of uric acid), kidney disease, pulmonary hypertension (high blood pressure in the arteries that supply the lungs), hepatic adenoma (benign liver tumors), polycystic ovaries in females, an inflammation of the pancreas (pancreatitis) and brain damage. Early diagnosis and effective treatment can result in normal growth and puberty and many affected individuals live into adulthood and enjoy normal life activities. Many female patients have had successful pregnancies.
Causes
Type I glycogen storage disease is associated with abnormalities in two genes. Mutations in the G6PC gene result in a deficiency in the glucose-6-phosphatase (G6Pase) enzyme and account for approximately 80% of GSDI. This type of GSDI is termed glycogen storage disease type Ia. Mutations in the SLC37A4 gene result in a deficiency in the glucose-6-phosphatase translocase enzyme (transporter deficiency) and account for approximately 20% of GSDI. This type of GSDI is termed glycogen storage disease type Ib. These enzyme deficiencies cause excess amounts of glycogen to be stored in the body tissues. In general GSD type Ib patients have similar clinical manifestations as type Ia patients but in addition, the white blood cells (neutrophils) are low in number and do not function normally (neutropenia). This can result in frequent infections and mouth ulcers.
Type I glycogen storage disease is inherited as an autosomal recessive genetic disorder. Recessive genetic disorders occur when an individual happens to inherit two copies of the same abnormal gene for the same trait/disease from each parent. If an individual receives one normal copy of the gene (allele) and one abnormal/defective copy of gene (allele) for the disease, the person will be a carrier for the disease, but usually will not show symptoms. Thus most parents of the person affected with GSD type I are unaffected carriers if the disease gene. The risk for two carrier parents having a child and passing the same defective gene to the new baby, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for GSDI is 25%. The risk is being affected is the same for males and females
Affected PopulationsType I glycogen storage disease occurs in approximately 1 in 100,000 births. The prevalence of GSDI in Ashkenazi Jews is approximately 1 in 20,000. This condition affects males and females in equal numbers in any given population group.
Related Disorders
Symptoms of the following disorders can be similar to those of glycogen storage disease type I. Comparisons may be useful for a differential diagnosis:
Forbes or Cori disease (GSD-III) is one of several glycogen storage disorders that are inherited as autosomal recessive traits. Symptoms are caused by a lack of the enzyme amylo-1,6 glucosidase (debrancher enzyme). This enzyme deficiency causes excess amounts of an abnormal glycogen (the stored form of energy that comes from carbohydrates) to be deposited in the liver, muscles and, in some cases, the heart. Symptoms become evident during the first years of life.
Andersen disease (GSD-IV) also known as glycogen storage disease type IV. This GSD is also inherited as an autosomal recessive trait. It is caused by deficient activity of the glycogen-branching enzyme (GBE), resulting in accumulation of abnormal glycogen in the liver, muscle, and/or other tissues. In most affected individuals, symptoms and findings become evident in the first few years of life. Such features typically include failure to grow and gain weight at the expected rate (failure to thrive) and abnormal enlargement of the liver and spleen (hepatosplenomegaly).
Hers disease (GSD-VI) is also called glycogen storage disease type VI and usually has milder symptoms than most other types of glycogen storage diseases. It is caused by a deficiency of the enzyme liver phosphorylase. Hers disease is characterized by enlargement of the liver (hepatomegaly), moderately low blood sugar (hypoglycemia), elevated levels of acetone and other ketone bodies in the blood (ketosis), and moderate growth retardation. Symptoms are not always evident during childhood, and children are usually able to lead normal lives. However, in some instances, symptoms may be severe.
Glycogen storage disease IX is caused due to deficiency of phosphorylase kinase enzyme (PK enzyme deficiency). It can be inherited as an X-linked genetic disorder caused by a deficiency of the enzyme liver phosphorylase kinase or it can be inherited as an autosomal recessive form causing liver and/or muscle disease. The disorder is characterized by slightly low blood sugar (hypoglycemia). Excess amounts of glycogen (the stored form of energy that comes from carbohydrates) are deposited in the liver, causing enlargement of the liver (hepatomegaly).
Standard Therapies
Treatment
GSDI is treated with a special diet in order to maintain normal glucose levels, prevent hypoglycemia and maximize growth and development. Frequent small servings of carbohydrates during the day must be maintained throughout life. Calcium, vitamin D and iron supplements may be recommended. Feeding of uncooked cornstarch is used to improve blood levels of glucose. Allopurinol, a drug capable of reducing the level of uric acid in the blood, may be useful to control the symptoms of gout-like arthritis during the adolescent years. Medications may be prescribed to lower lipid levels and prevent and/or treat kidney disease. Human granulocyte colony stimulating factor (GCSF) may be used to treat recurrent infections in type Ib patients. Liver tumors can be treated with surgery or a procedure in which current is used to heat and eliminate the tumor (radiofrequency ablation). Kidney and/or liver transplantation are sometimes considered if other therapies are unsuccessful or where liver adenomas keep growing.
Individuals with GSDI should be monitored at least annually with kidney and liver ultrasound and routine blood work specifically used for monitoring GSD patients.
Genetic counseling is recommended for affected individuals and their families.
REFERENCES FROM :
https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/
Glycogen Storage Disease Type INORD gratefully acknowledges Yuan-Tsong Chen, MD, PhD, Professor, Division of Medical Genetics, Department of Pediatrics, Duke Medicine; Director, Academia Sinica Institute of Biomedical Sciences, Taiwan and Deeksha Bali, PhD, Associate Professor & Director GSD Section, Biochemical Genetics Laboratories, Duke Medicine, for assistance in the preparation of this report.
